








Services on Demand
Journal

Article
 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkCiência e Cultura
Print version ISSN 0009-6725
Cienc. Cult. vol.63 no.1 São Paulo Jan. 2011
http://dx.doi.org/10.21800/S0009-67252011000100014

Os avanços tecnológicos na química analítica: sucessos e desafios
Quezia B. Cass
Juliana Cristina Barreiro
Quando pensamos em estratégias usadas no desenvolvimento de fármacos, notamos de imediato os avanços tecnológicos alcançados em todas as áreas da análise farmacêutica e biomédica. O desenvolvimento de uma nova entidade molecular bioativa envolve processos complexos e interdisciplinares, que demandam a participação de pesquisadores das mais diversas áreas em atuação conjunta: desde o químico de produtos naturais ao químico orgânico sintético, passando por farmacologistas, químicos medicinais, farmacêuticos, toxicologistas etc. Todos são essenciais nesse processo e todos trabalham em parceria com o químico analítico (1).
AS NOVAS PLATAFORMAS ANALÍTICAS O uso de pequenas moléculas tem sido fundamental para o entendimento dos processos biológicos. Os produtos naturais são coleções combinatórias com alta diversidade estrutural e têm sido usados com sucesso para pesquisa em biologia química. Na prospecção por moléculas bioativas, os avanços tecnológicos que proporcionaram a hifenação das técnicas de separação às técnicas de espectrometria de massa (MS) e ressonância nuclear magnética (NMR), permitem a identificação estrutural completa (ou parcial), on-line, de misturas complexas. Com isso, evita-se, portanto, a perda de tempo com isolamentos de compostos de baixo interesse estrutural e/ou de atividade biológica, além de se permitir a caracterização de compostos lábeis e/ ou voláteis de difícil isolamento (2-4).
No acoplamento LC-MS/MS, as interfaces de ionização a pressão atmosférica (API) – electrospray (ESI), ionização química a pressão atmosférica (APCI) e a fotoionização a pressão atmosférica (APPI) – permitem que sejam analisados compostos de todas as faixas de polaridade e massa molecular, além de tornar possível a seleção do modo de ionização. São, por isso, as mais empregadas no acoplamento com a LC para a análise quantitativa de compostos aquirais/quirais em matrizes complexas (5). O analisador do MS é, também, algo que deve ser cuidadosamente selecionado, pois elevada sensibilidade e seletividade são comumente alcançadas em múltiplos estágios (MS/MS) no tempo (a partir do uso de analisadores do tipo ion trap) e no espaço (com o uso de analisadores do tipo triplo quadrupolo (QqQ)) (6). O risco de falso positivo é significantemente reduzido com o uso de analisadores por tempo de voo (TOF), devido à elevada resolução e exatidão de massa alcançada sem que haja perda de sensibilidade. Analisadores híbridos, tais como quadrupo-lo-TOF (QTOF), têm sido utilizados atualmente para análises confirmatórias e elucidação estrutural de compostos em matrizes complexas (7; 8). Os avanços conseguidos por LC-API-MS permitiram o uso generalizado da técnica tanto para quantificação quanto para elucidação estrutural. Inicialmente, acreditava-se que o preparo de amostras poderia ser reduzido ao mínimo necessário. Entretanto, apesar da elevada sensibilidade e seletividade alcançadas, as análises feitas usando ESI e APCI são suscetíveis a efeitos de matriz; causados pela coeluição de componentes presentes na amostra, os quais afetam a ionização por supressão ou ganho na resposta do sinal. Alteram-se, portanto, a reprodutibilidade, a linearidade e a exatidão do método (9-11). A interface APPI é apontada como menos suscetível a efeito matriz que as interfaces ESI e APCI quando submetidas às mesmas condições de análises. O uso de nanoLC hifenado com espectrômetro de massas com ionização direta de elétrons (EI) é considerado uma alternativa à API para resolver efeito matriz (12).
NMR é indispensável em elucidação estrutural completa. Tem grande aplicação em determinação/caracterização de impurezas, metabolitos, extratos de produtos naturais, produtos de síntese etc. Como detector em LC, o fato de não ser destrutivo compensa sua baixa sensibilidade. Espectros de alta resolução são conseguidos pelo uso de extração em fase sólida (SPE) como interface ao NMR, o que permite o uso de solventes não deuterados para a separação cromatográfica. O uso de pequenos volumes de solvente deuterados (30–120 µl) é feito somente para transferência dos analitos para o probe, o que torna o processo de supressão de solvente para o espectro de NMR usualmente desnecessário (13).
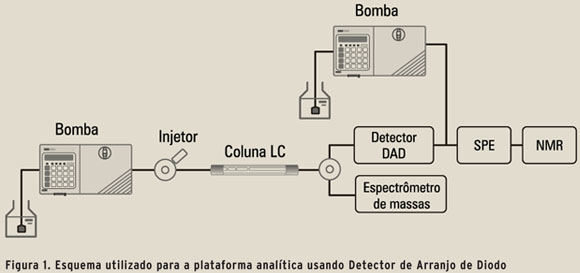
LC-MS-SPE-NMR é a plataforma analítica ideal para quantificação e identificação estrutural molecular de misturas complexas (14; 15). Embora ainda seja relativamente pouco utilizada, os desenvolvimentos recentes, tanto em MS quanto em NMR, indicam um crescimento na sua utilização (13; 14; 16). A Figura 1 mostra o esquema utilizado para essa plataforma usando o Detector de Arranjo de Diodo (DAD) para monitorar o efluente da coluna.
A capacidade de aliar as técnicas de separação/identificação estrutural a métodos de bioensaios permite a identificação de compostos já conhecidos e de novas moléculas de interesse em coleções combinatórias naturais e/ou sintéticas e tem sido explorada no âmbito de desenvolvimento de novos ensaios em massa (17). Métodos multivariados de tratamento de dados têm sido necessários e utilizados cada vez mais em processos analíticos complexos (18-20). A quimiometria tem sido utilizada em desenvolvimento de métodos, tratamento estatístico dos resultados analíticos encontrados e, especialmente, em modelos de padrão de reconhecimento (21).
QUÍMICA ANALÍTICA MEDICINAL Os avanços na área de química analítica medicinal (1) têm sido notórios e as novas plataformas tecnológicas de análise têm criado não só soluções, mas novos paradigmas e demandado maior desenvolvimento analítico.
Os ensaios em massa, por exemplo, exigem soluções criativas (22) para caracterização dos compostos em mistura que se liguem ao alvo selecionado. As demandas por automação, alta frequência analítica, baixos limites de quantificação, necessidade de menor manuseio de amostra e redução de resíduos químicos e biológicos nas análises têm, todas elas, exigido avanços nos processos de preparo de amostras. Merecem destaque aqueles que envolvem injeção direta de matrizes nativas (23; 24).
Os avanços em LC (nano, capilar, rápida e de ultraeficiência), associados à detecção por espectrometria de massa, têm fornecido plataformas tecnológicas eficientes, com a frequência analítica requerida para uma variedade de aplicações em bioanálise e em identificação estrutural de metabolitos (5; 25).
A utilidade de se usar processos metabolômicos (i) para se entender doenças deu aos pesquisadores da área um imenso desafio no que concerne ao desenvolvimento de ferramentas analíticas apropriadas para o manuseio, frequência analítica e tratamento de dados. A cromatografia líquida em duas dimensões 2DLC, do tipo abrangente (ii) (LCXLC), tem sido aplicada, majoritariamente, em estudos de proteomas (iii) e metabolomas, com o intuito de se obter o maior número de picos em menor tempo de análise, algo que não é possível, para os números hoje demandados, em uma dimensão. Além da maior capacidade de pico em menor tempo de análise, tem-se também maior eficiência analítica (resolução). É, portanto, uma ferramenta analítica poderosa em bioanálise. No entanto, a maioria dos equipamentos utilizados em LCXLC é ainda do tipo home-made e o tratamento dos dados obtidos ainda é um problema a ser solucionado com o uso de softwares apropriados (26; 27). Isso tem limitado as aplicações em análises farmacêuticas, biomédicas e biotecnológicas, embora análises por 2DLC de produtos de degradação tenham sido reportadas (28). Muitas vezes, o ganho em sensitividade obtido em MS após uma separação 2D é o resultado da pureza da banda cromatográfica, que evita, assim, a supressão da ionização (15). De tal sorte, espera-se que os equipamentos e softwares comerciais atendam à demanda da técnica.
A IMPORTÂNCIA DA ESTEREOSSELETIVIDADE Embora há muito se atribuísse grande importância à estereoquímica para sistemas bioquímicos e químicos em condições não isotrópicas (29), a síntese de compostos enantiomericamente puros só foi seriamente considerada quando se tornou viável a sua produção em larga escala. Os progressos em sínteses assimétricas, combinando abordagens biotecnológicas com novos desenvolvimentos em catálise assimétrica e novos métodos de resolução, facilitam a produção de tais compostos (30-32).
As aplicações de métodos para separação de enantiômeros estão esboçadas no Esquema 1

O desenvolvimento de métodos analíticos para resolução de enantiômeros criou um novo conceito no desenvolvimento de fármacos. Diante disso, os órgãos regulatórios passaram a exigir: as justificativas para a forma estereoquímica escolhida, a descrição da síntese assimétrica com método analítico para determinação da razão/pureza enantiomérica e a configuração absoluta do ativo. Também são exigidos métodos para estudos farmacocinéticos dos enantiômeros em separado (33; 34).
As dificuldades inerentes à separação de enantiômeros explicam o desenvolvimento tardio nos métodos cromatográficos de separação. Assim, não é de todo descabido o famoso chiste de que a separação de enantiômeros continua a ser uma arte, como nos tempos de Pasteur (35).
As fases estacionárias quirais para LC, lançadas comercialmente no início dos anos 1980, revolucionaram os métodos analíticos de resolução de enantiômeros e estabeleceram a cromatografia líquida como principal técnica de separação.
As misturas enantioméricas são resolvidas por meio de complexos diastereoisoméricos transitórios (iv) analito/fase estacionária quiral, que envolvem várias interações simultâneas no processo de discriminação quiral.
Uma variedade imensa de fases estacionárias quirais é comercialmente disponível. Elas são classificadas como pertencentes a dois grandes grupos de seletores quirais: naturais e sintéticos (36).
Dentre os naturais, destacam-se os derivados de polissacarídeos, de glicopeptídeos macrocíclicos, de proteínas e de ciclodextrinas. Dos sintéticos, merecem destaque os do tipo Pirkle, os de polímeros impressos molecularmente (MIPs) e os de polímeros sintéticos. A capacidade de se trabalhar no modo analítico e em escala multimiligrama ou preparativa é, talvez, responsável pelo grande sucesso da LC quando comparado à eletroforese capilar ou à cromatografia gasosa (GC), ambas também extensivamente usadas na separação de misturas enantioméricas (37).
Quando um fármaco é administrado como mistura racêmica, os parâmetros farmacocinéticos são mais complexos do que quando na forma enantiomericamente pura. Os enantiômeros podem ter Tmax e Cmax diferentes, devido à discriminação quiral sofrida nos processos farmacocinéticos. A razão enantiomérica (v) pode ainda ser afetada pela via de administração ou pelo sexo, idade, estado de saúde e fenótipo do paciente (38).
Apesar disso, antes da revisão do artigo "Sophisticated nonsense in pharmacokinetics and clinical pharmacology", de Ariens em 1984 (39), a importância da estereoquímica no cenário farmacêutico era ignorada. Hoje em dia, os estudos de bioequivalência (vi) ainda não usam métodos enantiosseletivos, embora as razões para a importância destes sejam já bem conhecidas (40).
Os métodos de isolamento de enantiômeros em grande escala, especialmente por meio da cromatografia de leito móvel simulado, têm favorecido a produção de enantiômeros puros para os estudos farmacológicos e ou toxicológicos (41).
Modelar em condições analíticas o escalonamento das separações preparativas com alta produtividade tem propiciado plataformas minituarizadas capazes de fazer previsões exatas em uma escala de até 1 milhão de vezes, resultando em redução de solvente, de acordo com as demandas tecnológicas da química analítica verde (42; 43).
Ainda no contexto da consciência ambiental, os avanços tecnológicos despertaram a atenção para a ocorrência de resíduos de fármacos ativos, provenientes da excreção humana e animal no meio ambiente (44). Como um grande número de fármacos comercializados é quiral (vii) e usado na forma de mistura racêmica, ou enantiomericamente pura, diferenças na toxicidade e disponibilidade de um enantiômero em relação ao outro podem existir, alterando a fração enantiomérica (45-47) (viii). O desenvolvimento de métodos analíticos (48) que propiciem a separação e quantificação de enantiômeros em nível traço, principalmente em sistemas aquáticos, surge como uma importante ferramenta, uma vez que possibilitam a investigação enantiosseletiva e monitoramento dos processos biológicos, tais como bióticos e abióticos, envolvidos durante a permanência dessas substâncias no ambiente (45; 49-51).
UM DOS MAIORES DESAFIOS Na formação de recursos humanos, o maior desafio é formar um indivíduo multidisciplinar, com sólido conhecimento em química fundamental, que lhe propicie as condições para atuar nas mais diversas áreas do conhecimento. A adequação na formação científica dos estudantes precisa ser revista, para englobar as demandas da química analítica moderna.
Quezia B. Cass é professora associada do Departamento de Química da Universidade Federal de São Carlos (UFSCar), pesquisadora do 1C do CNPq, coordenadora do grupo de pesquisa Síntese Orgânica e CLAE. Os interesses de pesquisa estão relacionados com desenvolvimento de métodos para quantificação de pequenas moléculas em matrizes nativas complexas. Email: quezia@cnpq.br
Juliana Cristina Barreiro é bolsista de pós-doutoramento da Fapesp no grupo de pesquisa Síntese Orgânica e CLAE do Departamento de Química da UFSCar.

REFERÊNCIAS BIBLIOGRÁFICAS
1. Espada, A.; Molina-Martin, M.; Dage,J.; Kuo, M. S. Drug Discovery Today, Vol.13, no.417. 2008.
2. Hostettmann,K.; Wolfender, J.-L.; Terreaux, C. Pharmaceutical Biolology, Vol.39, no.18. 2001.
3. Queiroz, E. F.; Wolfender, J. L.; Atindehou, K.K.; Traore, D.; Hostettmann, K. Journal of Chromatography A, Vol.974, no.123. 2002.
4. Li,D.Q.;QianZ.M.;Li,S.P.Journal of Agricultural and Food Chemistry, Vol.58, no.6608. 2010.
5. Holcapek, M.; Kolarova, L.; Nobilis, M. Analytical and Bioanalytical Chemistry, Vol.391, no.59. 2008.
6. Gosetti, F.; Mazzucco, E.; Zampieri, D.; Gennaro, M. C. Journal of Chromatography A, Vol.1217, no.3929. 2010.
7. Sancho,J. V.; Pozo, O. J.; Ibanez, M.; Hernandez, F. Analytical and Bioanalytical Chemistry, Vol.386, no.987. 2006.
8. Hernandez, F.; Sancho, J.V.; Ibanez, M.; Grimalt, S. Trac-Trend in Analytical Chemistry, Vol.27, no.862. 2008.
9. Cassiano, N.M.; Barreiro, J.C.; Martins, L.R.R.; Oliveira, R.V.; Cass, Q.B. Química Nova, Vol.32, no.1021. 2009.
10. Cappiello, A.; Famiglini, G.; Palma, P.; Trufelli, H. Journal of Liquid Chromatography & Related Technologies, Vol.33, no.1067. 2010.
11. Chambers, E.; Wagrowski-Diehl, D.M.; Lu, Z.L.; Mazzeo, J.R. Journal of Chromatography B, Vol.852, no.22. 2007.
12. Trufelli, H.; Palma, P.; Famiglini, G.; Cappiello, A. Mass Spectrometry Reviews (no prelo). 2010.
13. Tang, H.R.; Xiao, C.N.; Wang, Y.L. Magnetic Resonance in Chemistry, Vol.47, S157. 2009.
14. Corcoran, O.; Spraul, M. Drug Discovery Today. Vol.8, no.624. 2003.
15. Guttman, A.; Varoglu, M.; Khandurina, J. Drug Discovery Today, Vol.9, no.136. 2004.
16. Kuhnle, M.; Holtin, K.; Albert, K. Journal of Separation Science, Vol.32, no.719. 2009.
17. Calleri, E. et al. Journal of Medicinal Chemistry, Vol.53, no.3489. 2010.
18. Daolio, C. et al. Phytocheical Analysis, Vol.19, no.218. 2008.
19. Williams, R.E.; Lenz, E.M.; Lowden, J.S.; Rantalainen, M.; Wilson, I.D. Molecular Biosystems, Vol.1, no.166. 2005.
20. Williams, R.E.; Lenz, E.M.; Rantalainen, M.; Willson, I.D. Molecular Biosystems, Vol.2, no.193. 2006.
21. Lavine, B.; Workman, J. Analytical Chemistry, Vol.82, no.4699. 2010.
22. Ozbal, C.C. et al. Assay and Drug Development Technologies, Vol.2, no.373. 2004.
23. Cassiano, N.M.; Barreiro, J.C.; Moraes, M.C.; Oliveira, R.V.; Cass, Q. B. Bioanalysis, Vol.1, no.577. 2009.
24. Temporini, C.; Calleri, E.; Cabrera, K.; Felix, G.; Massolini, G. Journal of Separation Science, Vol.32, no.1120. 2009.
25. Guillarme, D.; Ruta, J.; Rudaz, S.; Veuthey, J.L. Analytical and Bioanalytical Chemistry, Vol.397, no.1069. 2010.
26. Stoll, D.R. Bioanalysis, Vol.2, no.105. 2010.
27. Stoll, D.R. Analytical and Bioanalytical Chemistry, Vol.397, no.979. 2010.
28. Alexander, A.J.; Ma, L.J. Journal of Chromatography A, Vol.1216, no.1338. 2009.
29. Kostyanovsky, R.G. Mendeleev Communications, Vol.13, no.85. 2003.
30. Legros, J.; Dehli, J.R.; Bolm, C. Advanced Synthesis & Catalysis, Vol.347, no.19. 2005.
31. Rouhi, A.M. Chemical & Engineering News, Vol.80, no.43. 2002.
32. Patel, R.N. Coordination Chemistry Reviews, Vol.252, no.659. 2008.
33. Marzo, A.; Heftmann, E. Journal of Biochemical and Biophysical Methods, Vol.54, no.57. 2002.
34. Waldeck, B. Pharmacology & Toxicology, Vol.93, no.203. 2003.
35. Lough, W.J.; Wainer, I.W. Chirality in natural and applied science. Blackwell Science; CRC Press, Oxford Boca Raton, pp. xiii, 313p. 2002.
36. Berthod, A. Analytical Chemistry, Vol.78, no.2093. 2006.
37. Ward, T.J.; Ward, K.D. Analytical Chemistry, Vol.82, no.4712. 2010.
38. Rentsch, K. M. Journal of Biochemical and Biophysical Methods, Vol.54, no.1. 2002.
39. Ariens, E.J. European Journal of Clinical Pharmacology, Vol.26, no.663. 1984.
40. Torrado, J.J.; Blanco, M.; Farre, M.; Roset, P.; Garcia-Arieta, A. European Journal of Clinical Pharmacology, Vol.66, no.599. 2010.
41. Junior, I. J. D. et al. Química Nova, Vol.29, no.1027. 2006.
42. Welch, C.J. et al. Trac-Trend in Analytical Chemistry, Vol.29, no.667. 2010.
43. Armenta, S.; Garrigues, S.; De la Guardia, M. Trac Trends in Analytical Chemistry, Vol.27, no.497. 2008.
44. MacLeod, S.L.; Wong, C.S. Water Research, Vol.44, no.533. 2010.
45. Hashim, N.H.; Shafie, S.; Khan, S.J. Environmental Technology, Vol.31, no.1349. 2010.
46. Fono, L.J.; Sedlak, D.L. Environmental Science & Technology, Vol.39, no.9244. 2005.
47. Buser, H.R.; Poiger, T.; Muller, M.D. Environmental Science & Technology, Vol.33, no.2529. 1999.
48. Barreiro, J.C.; Vanzolini, K.L.; Madureira, T.V.; Tiritan, M.E.; Cass, Q.B. Talanta, Vol.82, no.384. 2010.
49. Matamoros, V.; Hijosa, M.; Bayona, J.M. Chemosphere, Vol.75, no.200. 2009.
50. Ali, I.; Singh, P.; Aboul-Enein, H.Y.; Sharma, B. Analytical Letters, Vol.42, no.1747. 2009.
51. Perez, S.; Barcelo, D. Trac-Trend in Analytical Chemistry, Vol.27, no.836. 2008.