









Servicios Personalizados
Revista

Articulo
 Portugués (pdf)
Portugués (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkCiência e Cultura
versión On-line ISSN 2317-6660
Cienc. Cult. vol.69 no.3 São Paulo jul./set 2017
http://dx.doi.org/10.21800/2317-66602017000300009
ARTIGOS
INOVAÇÃO E TRANSFORMAÇÃO
Cristalografia macromolecular: a biologia sob a ótica dos raios X
André Luis Berteli AmbrosioI; Kleber Gomes FranchiniII
IPesquisador vinculado ao Laboratório Nacional de Biociências (LNBio), junto ao Centro Nacional de Pesquisas em Energia e Materiais (CNPEM)
IIProfessor titular do Departamento de Clínica Médica da Universidade Estadual de Campinas (Unicamp) e diretor do Laboratório Nacional de Biociências (LNBio), associado ao Centro Nacional de Pesquisa em Energia e Materiais (CNPEM)
Um décimo de milímetro. Essa é a menor distância resolvível pelo olho humano, limitada em grande parte pela anatomia da pupila e pela separação das células fotorreceptoras na retina. Esse nível de acuidade visual, combinado com a capacidade de distinguir objetos com base na luz que refletem, emitem ou transmitem (nos comprimentos de onda entre 400 e 700 nanômetros) foi capaz de sustentar a sobrevivência do Homo sapiens anatomicamente moderno nos últimos 200 mil anos (1). Enquanto questões sobre a origem, o funcionamento e as estruturas dos seres vivos têm sido perseguidas por praticamente todas as culturas conhecidas ao longo da história, somente há pouco mais de três séculos - após o advento de lentes e microscópios - é que fomos capazes de compreender que não somente nós, mas também todos os outros organismos vivos são formados por blocos básicos chamados células, produzindo uma compreensão notavelmente detalhada de como os seres vivos operam. Esta nova compreensão, que chamamos coletivamente de biologia molecular, foi, e ainda é, fundamentada na física e na química.
Hoje, sabemos que células proveem a estrutura organizacional dentro das quais milhares de transformações químicas acontecem em escalas de tempo extremamente curtas, permitindo que os seres vivos cresçam, se adaptem ao ambiente, respondam a estímulos externos e se reproduzam. Células medem tipicamente alguns poucos micrômetros, ou seja, por volta de duas ordens de magnitude abaixo da capacidade natural da visão humana. Ainda assim, o interior da célula é um ambiente funcionalmente bem organizado, densamente ocupado e dinâmico, o que o torna fascinante. Para se ter uma ideia, o núcleo de uma célula humana - cujo volume é dezenas de bilhões de vezes menor que o de uma gota de chuva - acomoda praticamente dois metros de material genético condensado. Estima-se que o corpo de uma pessoa adulta seja formado por cerca de 30 trilhões de células especializadas, das quais apenas 20% carregam DNA (2). Porém, ainda que fosse possível esticar todo o DNA desses seis trilhões de células, teriam que ser realizadas 80 viagens de ida e volta entre a Terra e o Sol.
Seja uni ou multicelular, para cada tarefa em um organismo existe uma classe de moléculas, coletivamente chamadas de proteínas, destinada a realizá-la. Proteínas são responsáveis por empacotar o material genético dentro do núcleo, por permitir sensibilidade à luz, distinção entre cores, cheiros e sabores. São também responsáveis pela adaptação ao meio ambiente, pelo transporte de oxigênio no sangue, pela realização de funções cerebrais conscientes e subconscientes, pela contração muscular, pela conversão de alimentos em energia ou mesmo por produzir novas proteínas e células. Portanto, proteínas são complexas máquinas moleculares que desempenham todas as funções fundamentais que sustentam a vida. Eventuais desregulações na execução dessas funções, portanto, podem levar ao desenvolvimento de doenças como, por exemplo, o câncer, o que transforma as proteínas em alvos da maioria dos medicamentos hoje disponíveis.
O PARADIGMA FORMA-FUNÇÃO
Do ponto de vista químico, proteínas são polímeros formados pela associação linear de centenas ou milhares de resíduos de aminoácidos. Apesar de somente 20 tipos de aminoácidos serem encontrados nas células, pelo menos 32 mil proteínas distintas são produzidas em humanos, segundo estimativas conservadoras (3), nas mais variadas combinações sequenciais. As receitas precisas para as proteínas são codificadas pelo DNA.
Hoje, entendemos que há uma relação íntima entre a estrutura tridimensional das proteínas e a sua função. A disposição de cada um dos átomos - a sequência linear primária - define o enovelamento resultante e, portanto, a forma da proteína, que por sua vez determina a sua ação. Cada enovelamento é conduzido por uma série de interações não-covalentes, tais como ligações de hidrogênio, interações iônicas, forças de Van der Waals e empacotamento hidrofóbico. Estruturas atômicas ajudam a entender como as proteínas funcionam e, de maneira importante, como encontrar ou projetar pequenos compostos químicos que se encaixem de maneira a modular seletivamente as funções das proteínas. Mais precisamente, uma vez que uma proteína é identificada como um promissor alvo terapêutico, o estudo estrutural sobre a ligação de uma molécula de fármaco mostrará como o composto interage com a proteína. Esse conhecimento fornece bases sólidas para as sugestões de como um potencial fármaco pode ser modificado visando melhorar as propriedades de ligação e, também, onde não pode ser modificado para que a ligação não seja impedida.
Existe um ramo da ciência dedicado a entender quais são e como as proteínas adquirem suas estruturas e também como alterações nessas estruturas afetam sua função. Essa área é conhecida como biologia estrutural e agrega técnicas complementares de fronteira, como cristalografia por difração de raios X, ressonância magnética nuclear, crio-microscopia eletrônica, espectrometria de massas, entre outras. Todavia, dentro do estado da arte atual, cristalografia por difração de raios X ainda é o método mais utilizado e avançado para obter informações estruturais de alta resolução sobre macromoléculas biológicas.
Em linhas gerais, a cristalografia é rotineiramente usada para a obtenção de dados de difração de raios X de cristais únicos, a temperaturas criogênicas (100 K), onde os danos por radiação são atenuados. Em casos de sucesso, permite a construção de modelos moleculares precisos, compostos de dezenas de milhares de átomos, com separação visual de ordens de tamanho menores que os de uma ligação covalente (~ 1Å). As estruturas biológicas mais importantes foram obtidas com base em estudos de difração de raios X: a dupla hélice do DNA, inúmeras proteínas, vitaminas e fármacos. Atualmente, centenas de milhares de estruturas já foram determinadas por essa técnica.
CRISTALOGRAFIA POR DIFRAÇÃO DE RAIOS X
Diferentemente das células e seus subcompartimentos, proteínas são moléculas muito pequenas para que possamos vê-las sob microscopia de luz visível. Uma célula vermelha sanguínea humana ocupa um volume típico de 90 fentolitros (três bilhões de vezes menor que uma gota de chuva) e, ainda assim, pode ser facilmente vista através de um microscópio ótico. Um terço do seu volume é ocupado por cerca de 270 milhões de moléculas de hemoglobina (4), que são proteínas de aproximadamente 9000 átomos cada, responsáveis pelo transporte de oxigênio da respiração.
A limitação no uso da radiação visível na visualização da estrutura de proteínas se deve, principalmente, ao fato do comprimento de onda correspondente (centenas de nanômetros) ser muito maior do que o da molécula estudada (nanômetros) e, também, do nível de detalhes que pretendemos enxergar: seus átomos constituintes e as ligações entre eles (um décimo de um bilionésimo de metro). A radiação eletromagnética apropriada precisa ser compativelmente muito menor. Utilizamos, portanto, raios X, que não conseguimos detectar diretamente com nosso olho.
Porém, um experimento típico de "visualização" de estruturas de proteínas utilizando raios X não é, em princípio, conceitualmente diferente do mecanismo da visão humana: necessitamos de uma fonte de luz cuja radiação emitida interagirá com o objeto de interesse em nível quântico, excitando elétrons em átomos através de absorção, emissão e espalhamento. O resultado dessa interação é então detectado por um sistema apropriado, a partir do qual a informação é processada e interpretada (nesse caso matematicamente) à forma do objeto estudado.
Os comprimentos de onda de raios X utilizados para a determinação da estrutura de proteínas são tipicamente da ordem de 1 Å (10-10 m). Nas energias correspondentes (~12 keV), praticamente 99% da radiação atravessa a amostra sem interagir com os átomos. Apenas uma fração mínima (< 10%) dos fótons que de fato interagem com a amostra serão espalhados elasticamente pelos átomos desta, carregando assim informações estruturais úteis. A detecção desse sinal, geralmente muito fraco, é um desafio por si, que não é simplesmente resolvido ajustando-se para longas exposições. O restante dos fótons que interagem com os átomos depositará energia na amostra (devido ao efeito fotoelétrico, emissão Auger e espalhamento inelástico), causando ionização e aquecimento e, portanto, danificando rapidamente as moléculas de maneira irreversível.
Devido a limitações experimentais intrínsecas, relacionadas principalmente ao baixo brilho de fontes de raios X e à baixa sensibilidade dos sistemas de detecção da radiação espalhada (filmes fotográficos, luminescência foto-estimulada etc.), a solução encontrada para a amplificação do sinal relevante de espalhamento de uma única molécula - que seria outrora incomensurável - foi a utilização de cristais.
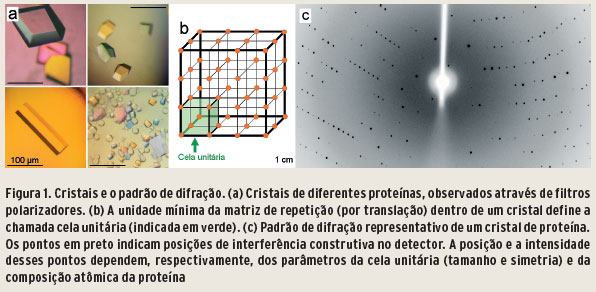
Em determinadas condições químicas de solubilidade (supersaturação), moléculas individuais de proteína podem ser levadas a formar cristais (Figura 1a-b), empacotando-se aos bilhões em uma matriz de repetição e mantidas unidas por interações não covalentes. Proteínas variam consideravelmente entre si em parâmetros físico-químicos (tamanho, carga líquida, forma), de maneira que a determinação das condições ideais para a formação de cristais é empírica. Em casos de sucesso, o processo requer extensa variação de parâmetros químicos de soluções combinados, como força iônica, pH e temperatura, entre outros.
Ao serem iluminados por um feixe colimado de raios X, cristais se comportam como uma grade de difração tridimensional, o que dá origem a efeitos de interferência construtiva e destrutiva. Os eventos de interferência construtiva aparecem no detector como uma série de pontos discretos, que são conhecidos como reflexões (Figura 1c). Em um experimento típico, milhares de reflexões são medidas simultaneamente. No caso do espalhamento elástico, o campo elétrico oscilante da radiação incidente faz com que os componentes carregados dos átomos oscilem com a mesma frequência. Cada dipolo oscilante retorna a um estado menos energético, emitindo um fóton com a mesma energia que a energia dos fótons incidentes. A intensidade dispersa é inversamente proporcional ao quadrado da massa da partícula que emite o fóton de raio X. Pelo fato de ser muito pesado, a dispersão de raios X pelo núcleo é considerada desprezível em comparação com a dispersão pelos elétrons de um átomo. Assim, considera-se que o espalhamento dos raios X é devido à densidade eletrônica dos átomos em uma proteína.
Como toda forma de radiação eletromagnética, raios X têm propriedades de onda, ou seja, podem ser descritos por uma amplitude e uma fase, associadas à respectiva frequência de oscilação. Para recombinar matematicamente um padrão de difração e assim calcular a distribuição espacial de densidade eletrônica (promediada no espaço no cristal) da proteína estudada, ambos os parâmetros - amplitude e fase - são necessários para cada uma das reflexões medidas.
A intensidade da reflexão medida é proporcional ao quadrado da amplitude do fóton espalhado, bem como à energia transferida por unidade de área do detector durante um intervalo de tempo de exposição à radiação. A exposição típica é da ordem de milisegundos, e determinada principalmente pela sensibilidade do detector. Tal intervalo de tempo é, todavia, muito acima do período de oscilação da radiação incidente (exahertz): em um milissegundo, aproximadamente 1015 oscilações do fóton incidiram no detector. Portanto, enquanto a amplitude da reflexão pode ser diretamente estimada a partir de sua intensidade, toda informação da fase deste fóton é sistematicamente perdida durante a coleta. Esse fenômeno, historicamente conhecido como "o problema das fases", constitui uma limitação fundamental da técnica.
Felizmente, no caso da difração de cristais de macromoléculas biológicas, existem três possibilidades distintas de soluções experimentais para o problema das fases, sendo que cada uma apresenta aplicabilidade, desafios e limitações práticas inerentes.
A solução originalmente desenvolvida, chamada de substituição isomórfica, consiste na adição deliberada (derivatização) de átomos eletronicamente mais densos - como mercúrio, platina, chumbo e urânio - nos cristais de proteínas. A interação ordenada e consistente desses metais com regiões específicas das proteínas dentro do cristal altera de maneira significativa as intensidades das reflexões no padrão de difração. A partir da comparação das intensidades derivatizadas com as de cristais não-derivatizados, é possível inferir com precisão a posição dos átomos "pesados" e estimar um conjunto inicial de fases para todas as reflexões.
Outra alternativa para a solução do problema das fases consiste na chamada dispersão anômala e se baseia na variação do comprimento de onda do raio X incidente. Se a energia equivalente corresponde a uma transição entre diferentes camadas de elétrons de um tipo de átomo específico, haverá uma modificação das fases da onda difratada devido à absorção de fótons incidentes. Os átomos relativamente leves encontrados em proteínas, como carbono, nitrogênio e oxigênio não contribuem para a dispersão anômala em comprimentos de onda de raios X normais utilizados para a cristalografia de raios X (entre 0,7 e 2 Å). Assim, para se observar esse efeito, o átomo pesado deve ser nativo da proteína (enxofre, por exemplo) ou derivatizado, como no caso da substituição isomorfa. Além disso, o comprimento de onda do raio X emitido pela fonte deve ser ajustado às bordas de absorção do átomo pesado, geralmente correspondentes a energias das camadas eletrônicas mais internas (K ou L).
Por último, quando um modelo atômico para uma proteína homóloga àquela que se está estudando já está disponível, determinado por uma das metodologias acima, ele pode ser usado como fonte para um conjunto inicial de fases. Com a suposição de que ambas as proteínas (a de interesse e a modelo) desempenham a mesma função em espécies diferentes e compartilham um ancestral comum, funções matemáticas de rotação e translação que correlacionam as posições atômicas no cristal podem ser calculadas. Esse método é chamado de substituição molecular e seu sucesso baseia-se no acúmulo de estruturas resolvidas.
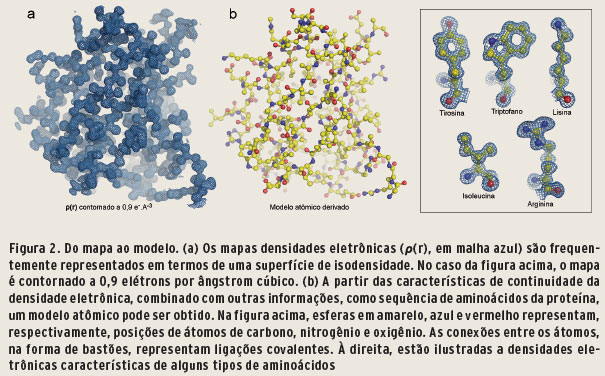
Quando cristalógrafos dizem ter resolvido uma estrutura, significa que o problema das fases foi resolvido. Em outras palavras, aplicando-se uma das três metodologias descritas acima, estimativas de fases suficientemente boas foram determinadas, de maneira a permitir o cálculo de um mapa de densidade eletrônica interpretável, que pode ser melhorado de forma iterativa, convergindo para um modelo estrutural fiel da proteína de interesse (Figura 2).
CONTEXTO HISTÓRICO
O campo da cristalografia por difração de raios X aplicado à biologia, como conhecemos hoje, surgiu do que foi provavelmente a primeira interação interdisciplinar de sucesso e talvez uma das mais revolucionárias na história da ciência moderna. A primeira metade do século XIX testemunhou o advento e junção de duas disciplinas até então distintas.
A primeira delas, pouco mais antiga, é a cristalografia - hoje definida como a ciência experimental dedicada à determinação da disposição de átomos nos sólidos cristalinos, mas que foi originalmente fundada pelo mineralogista francês René-Just Haüy, por volta de 1800, com base nos seus estudos sobre a geometria (hábito) de cristais inorgânicos. A segunda disciplina é a química orgânica, derivada dos trabalhos colaborativos de Jöns Jacob Berzelius e Gerardus Johannes Mulder, que cunharam o termo proteína e definiram suas primeiras composições químicas em 1838. Imediatamente depois, foi descoberto de maneira acidental que a hemoglobina (a proteína transportadora de oxigênio no sangue) poderia ser cristalizada quase que espontaneamente, a partir de amostras de sangue de virtualmente qualquer espécie animal. Com base nos estudos de minerais estabelecidos por Haüy, a caracterização morfológica sistemática dos primeiros cristais de moléculas biológicas se seguiu. Porém, o estudo de seus constituintes, em nível molecular, só seria possível com o advento de técnicas mais sofisticadas que surgiriam mais de sessenta anos depois.
Foi em 1912 que Max von Laue e colaboradores demonstraram que cristais inorgânicos, quando irradiados com raios X (então recentemente descobertos por Wilhelm Röntgen), eram capazes de espalhar essa radiação em diferentes direções, formando padrões de interferências construtivas e destrutivas. Logo em seguida, o jovem Laurence Bragg, então com 22 anos, percebeu que seria possível interpretar esse padrão de espalhamento matematicamente e assim determinar a estrutura atômica constituinte dos cristais. Com base nos experimentos realizados por seu filho, William Bragg construiu o primeiro espectrômetro de raios X, permitindo o posicionamento preciso das faces dos cristais com relação ao feixe de radiação e também a quantificação da intensidade dos raios X espalhados. Assim, a primeira estrutura cristalográfica, do cloreto de sódio (sal de cozinha), foi determinada. Max von Laue recebeu o prêmio Nobel em física em 1914 e os Bragg, pai e filho, foram laureados em 1915. No ano da premiação, Laurence Bragg tinha somente 25 anos, sendo ainda hoje o laureado mais jovem em qualquer uma das áreas contempladas.
A geração de cientistas que inicialmente se formou sob a tutela dos Bragg foi a que de fato desenvolveu a aplicação biológica da cristalografia. John Desmond Bernal, aluno de William Bragg, foi o pioneiro. Ele e sua estudante Dorothy Crowfoot Hodgkin produziram as primeiras e excelentes imagens de difração de uma proteína, a pepsina, em 1934. Apesar de o padrão de difração de cristais de proteínas ser extremamente mais complexo (em números de reflexões) do que aqueles de sais estudados por Bragg, estava claro para Bernal que esses padrões continham praticamente todas as informações necessárias para determinar a estrutura de uma proteína. Havia, porém, o problema das fases: a estrutura da pepsina não poderia ser imediatamente decifrada.
O problema das fases persistiu por mais de 20 anos após os resultados iniciais de Bernal e Hodgkin, até que Max Perutz (ex-aluno de Bernal) e colegas resolveram comparar padrões de cristais de proteínas contendo diferentes átomos de metais pesados. A partir dessas comparações, desenvolveram uma abordagem que deu origem ao método de substituição isomórfica. Em 1958, John Kendrew aplicou a técnica desenvolvida por Perutz, que foi seu orientador, para produzir as primeiras imagens tridimensionais de uma proteína: a mioglobina, que é utilizada pelos músculos para armazenar oxigênio.
Durante as décadas de 1920 e 1930, William Astbury, outro ex-aluno de William Bragg, foi pioneiro no uso de raios X para estudar fibras biológicas e no desenvolvimento dos métodos que seriam mais tarde utilizados por Rosalind Franklin e seu aluno Raymond Gosling, na obtenção da "foto 51". Esse é o apelido dado à famosa imagem de difração de raios X da dupla fita da forma B do DNA, usada controversamente como evidência experimental por Francis Crick (também aluno de Perutz) e James Watson na determinação da respectiva estrutura.
Nas décadas que se seguiram, dezenas de novas estruturas foram descritas por grupos independentes. Até hoje, todas as estruturas resolvidas são disponibilizadas à comunidade científica, por meio de repositórios universais especializados como, por exemplo, o Protein Data Bank, ou PDB, em www.rcsb.org. De acordo com as estatísticas de deposição do PDB, até o início da década de 1990, em torno de 500 estruturas de proteínas haviam sido determinadas. Esse número cresceu de maneira extraordinária nas décadas seguintes. No ano 2000, 13.600 estavam disponíveis e, em 2010, eram quase 70.000. Quando da escrita deste artigo, esse número havia ultrapassado a marca de 128.000.
O conhecimento da estrutura atômica de proteínas revolucionou o entendimento de uma ampla gama de mecanismos biológicos fundamentais tais como as bases moleculares da transcrição e tradução de genes, sinalização inter e intracelular, estruturação da membrana celular e estrutura e transporte através de poros transmembrana, infecção viral, interação patógeno-hospedeiro, catálise enzimática, entre outros inúmeros processos biológicos. Todo o entendimento em nível molecular do amplo espectro da biologia moderna se deve à enorme contribuição da cristalografia por difração de raios X.
Para se ter uma ideia de sua relevância, pelo menos 28 dos cientistas laureados no prêmio Nobel, até o momento, devem seu sucesso a algum aspecto do desenvolvimento ou da utilização da cristalografia por difração de raios X. Mesmo depois de mais de 100 anos desde seu desenvolvimento, o impacto dessa técnica na biologia ainda é extremamente relevante, graças, em grande parte, às instalações modernas, como o Sirius.
DESAFIOS, SOLUÇÕES TECNOLÓGICAS E O MARCO HISTÓRICO: FONTES SÍNCROTRON
Os passos e os desafios experimentais envolvidos na obtenção de estruturas cristalográficas são praticamente os mesmos desde a determinação das primeiras estruturas, na década de 1960. Avanços rápidos e contínuos na instrumentação permitiram, no entanto, que soluções fossem encontradas de maneira rápida, expandindo, principalmente, as fronteiras sobre quais tipos de proteínas poderiam ser estudas.
Por exemplo, para se identificar condições químicas que favoreçam o crescimento de cristais com qualidade suficiente para experimentos de difração, é necessária a produção de grandes quantidades da proteína de interesse, tipicamente na ordem de miligramas, com altíssimo grau de pureza e homogeneidade de espécies. No passado, as proteínas eram extraídas de fontes naturais, como plantas, microrganismos e tecidos, o que impunha limitações importantes ao tipo de proteína e de organismo fonte e também no rendimento da preparação. Com o advento de técnicas de biologia molecular (reação de polimerização de DNA em cadeia, técnicas de corte e ligação de porções de DNA, entre outros), o desenvolvimento de sistemas de expressão heterólogos (em bactérias ou leveduras, por exemplo) e culturas de células em larga escala, praticamente qualquer proteína, independente da origem, pode hoje ser produzida com alto rendimento, viabilizando assim a realização de tentativas iniciais de cristalização. Atualmente, proteínas humanas funcionalmente relevantes e de difícil extração podem ser estudadas estruturalmente.
A cristalização de macromoléculas biológicas ocorreu historicamente de maneira empírica. Nas últimas décadas, entretanto, a metodologia desenvolveu progressivamente uma maior racionalidade, como resultado de uma compreensão mais profunda dos processos físicos e químicos envolvidos. Com o advento de métodos automatizados (robôs) para manipulação dos cristais e montagem dos experimentos, vantagens cruciais foram obtidas em termos de miniaturização, precisão, velocidade e capacidade de trabalhar continuamente por períodos prolongados. Sistemas de visualização e identificação de cristais também são automatizados e se baseiam não somente em microscopia de luz visível, mas também em absorção e fluorescência na faixa de ultravioleta. Todavia, por mais que as chances de sucesso tenham sido consideravelmente aumentadas, a cristalização de macromoléculas biológicas ainda se baseia amplamente num processo de tentativa e erro.
Algumas das fontes originais de raios X mais utilizadas em cristalografia no passado foram variantes de tubos em vácuo, a partir dos quais a radiação eletromagnética é produzida por bombardeamento de ânodos rotatórios. Tais fontes eram geralmente suficientes para estudos dos cristais relativamente grandes (centenas de micrômetros) formados por moléculas de proteínas comparativamente pequenas (centenas de aminoácidos). Ainda assim, a coleta de dados levava muitos dias ou semanas. Originalmente, os cristais precisavam ser orientados manualmente com relação ao feixe, o que implicava em aumento considerável no tempo da coleta. No caso da obtenção da primeira estrutura da proteína lisozima, em 1967, foram necessários 14 dias consecutivos para que fosse coletado um único conjunto completo de alta resolução (5). No início da década de 1980, essas fontes passaram a incorporar os então recém desenvolvidos goniômetros de rotação e oscilação, permitindo controle automático da posição dos cristais. Uma grande limitação dessas chamadas "fontes caseiras" é o fato de que, ainda hoje, produzem radiação somente em um comprimento de onda, que é característico do material do qual o ânodo é feito. A radiação da transição K do cobre (λ = 1,54 Å) é quase que exclusivamente utilizada. Ânodos rotatórios de cromo (λ= 2,29 Å) e molibdênio (λ = 0,71 Å) também são utilizados, todavia de maneira bem menos frequente no estudo de cristais de proteínas.
Essa situação mudou radicalmente no início dos anos 1990, com a introdução de síncrotrons como poderosas fontes de radiação para a difração de cristais de macromoléculas biológicas. A característica principal dessa fonte é o alto brilho - com baixa divergência angular - do feixe de radiação produzido, através de um amplo espectro de energias, além de possibilitar a seleção precisa de comprimentos de onda.
Longos anéis de armazenamento em laboratórios síncrotron permitem a geração e estabilização de feixes de elétrons com emitância reduzida (a emitância é um parâmetro cinemático invariante em um acelerador, que caracteriza o tamanho do feixe de partículas). Em uma fonte síncrotron, quanto mais baixa a emitância do feixe de elétrons, melhor: pois menor e mais brilhante será o feixe resultante de raios X. O brilho é talvez o parâmetro mais importante da medida de qualidade e que determina a capacidade experimental de uma fonte de raios X. A evolução do brilho em função do progresso tecnológico na produção de raios X, usados no estudo de cristais de macromoléculas biológicas, é ilustrada na Figura 3a.
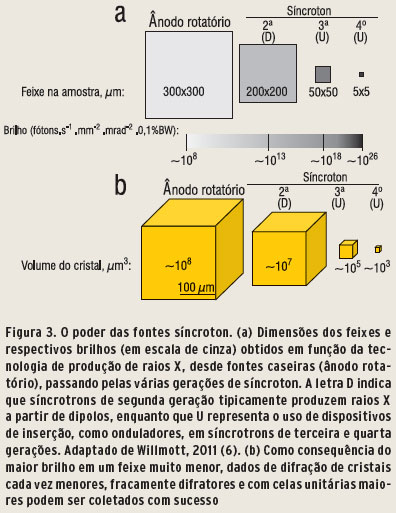
Em fontes caseiras, a grande seção de choque dos elétrons com o ânodo resulta em feixes de raios X de baixo brilho, que são compensados por uma menor colimação através de fendas. Tipicamente, o feixe que incide na amostra tem área de 90.000 µm2. Síncrotrons de segunda geração (como o LNLS, Campinas), com circunferências da ordem de uma centena de metros, permitem a produção de raios X oriundos de dipolo magnéticos. Através do uso de componentes óticos (espelhos e monocromadores), feixes com metade da área, cerca de mil vezes mais brilhantes que o de um ânodo, podem ser obtidos.
Síncrotrons de terceira geração são mais longos - com cerca de 500 metros de circunferência - e permitem emitância consideravelmente menor. As longas secções retas nesses anéis de armazenamento podem ainda acomodar dispositivos de inserção, tais como onduladores, que consistem numa longa disposição periódica de ímãs curtos (milímetros), com polaridade alternada. Os elétrons movem-se então em uma trajetória ondulada através desses ímãs, com deflexão zero de sua trajetória no anel de armazenamento, emitindo radiação extra por conservação de energia. No referencial relativístico do elétron, o pico da energia extra emitida é compatível com raios X e contribui significativamente com o aumento do brilho na amostra.
Portanto, um novo salto foi conseguido para síncrotrons de terceira geração, nos quais linhas com dispositivos de inserção com onduladores podem alcançar feixes de 2.500 µm2, três ordens de grandeza mais brilhantes que na geração anterior. Ainda assim, em síncrotrons de quarta geração, como o Sirius, principalmente através de tecnologia moderna, será possível fornecer aos usuários feixes com 25 µm2 ou menores, 10.000 vezes mais brilhantes.
Os métodos de coleta de dados de difração inicialmente envolviam filmes fotográficos com sensibilidade relativamente baixa. Eram necessárias longas exposições e quantificação manual e comparativa das intensidades das reflexões medidas. Scanners ópticos permitiram quantificações melhores e mais eficientes das reflexões. No entanto, o verdadeiro avanço foi a introdução de dispositivos automáticos controlados por computador, inicialmente com câmaras multifios, seguida por placas de imagem fosforescentes e câmeras CCD (dispositivos de carga acoplada).
Placas de imagens e CCD possuem uma ampla faixa dinâmica, boa linearidade de resposta, resolução espacial e grande área ativa (coletando dados de mais alta resolução), tudo isso com custo relativamente baixo. Porém, apresentam uma desvantagem que inviabiliza completamente seu uso em síncrotrons modernos, que é o tempo de leitura muito lento.
Hoje em dia, a alternativa compatível utilizada são os detectores ativos de pixels baseados em semicondutores de metal-óxido complementar (CMOS). Estes permitem a contagem direta e instantânea de fótons únicos de raios X. Devido a um tempo de leitura extremamente rápido (da ordem de microsegundos), os dados podem ser continuamente coletados à medida que o cristal é submetido à rotação; não há necessidade de oscilação de goniômetro ou mesmo de um obturador para delimitar os tempos de exposição individuais, como acontece com os sistemas de placas de imagens e CCD. Ainda, como importante consequência das altíssimas taxas de leitura (> 300 Hz), as diversas fontes de ruídos são praticamente eliminadas. Isso, combinado com alta faixa dinâmica, permite a importante quantificação de reflexões extremamente fracas (tipicamente a altas resoluções) e a consequente extensão da qualidade do modelo atômico obtido.
Os intensos feixes de raios X são capazes de danificar os cristais instantaneamente durante a coleta de dados. Esse processo indesejável pode ser retardado através da manutenção dos cristais a baixíssimas temperaturas. Normalmente, durante a coleta de dados, o cristal é mantido em nitrogênio gasoso a 100 K (-173,15 ºC). Por mais que a ionização e consequente formação de radicais livres ainda aconteça, a difusão destes através dos canais de solvente dos cristais é mitigada. Hoje em dia, apenas um cristal é tipicamente necessário para a coleta de um conjunto completo de dados de difração.
A coleta sob condições criogênicas trouxe outro benefício: os usuários podem congelar e armazenar os cristais com antecedência. Eles podem ser transportados durante dias, em distâncias intercontinentais, sem maiores complicações. Isso, combinado com a automação de passos envolvidos no posicionamento do cristal ante o feixe, permite o acesso do usuário a fontes de luz síncrotron distantes. Em diversas estações experimentais, é possível ao usuário somente enviar os cristais congelados, conduzindo a coleta dos dados em sua instituição de origem, via internet. Um dos benefícios imediatos é a economia de recursos financeiros que outrora seriam dedicados a pagar despesas de traslado (muitas vezes entre países) e estadia.
Outras vantagens relevantes de modernas estações experimentais em síncrotrons são: (i) devido ao seu tamanho, essas enormes fontes podem produzir feixes de luz que são pelo menos dez vezes mais finos que o diâmetro do cabelo humano. Assim, é possível varrer microvolumes dentro de cristais maiores, buscando detectar regiões mais ordenadas e, portanto, a melhor porção do cristal para difração; (ii) A capacidade de se ajustar a energia da radiação permite que os comprimentos de onda sejam precisamente selecionados para explorar a dispersão anômala de uma grande quantidade de elementos químicos, facilitando assim a obtenção de fases experimentais para determinação das estruturas. Nas novas fontes, pretende-se estender consideravelmente entre 0,3 e 5 Å de comprimento de onda; (iii) goniômetros modernos com vários graus de liberdade permitem a cobertura de toda a esfera de difração, com precisão de segundos de grau; (iv) possibilidade de difração in situ, onde cristais são testados ainda nas placas de cristalização.
Comparativamente, a coleta de um conjunto completo de dados levava dias em geradores "caseiros" (ânodos rotatórios) e horas e minutos em linhas síncrotrons de segunda e terceira gerações, respectivamente. No Sirius (fonte de quarta geração), apenas alguns poucos segundos serão suficientes.
Em termos gerais, podemos estimar que uma coleta de dados para um cristal gere aproximadamente 43 GB de dados em menos de 15 segundos (rotação de 360º em intervalos de 0,1º, 300 imagens por segundo, 12 MB por imagem). Portanto, a infraestrutura computacional de transferência, armazenamento e processamento de dados precisa andar de mãos dadas com a capacidade das estações experimentais. Novos algoritmos e programas de computação para a coleta de dados de difração, solução da estrutura e refinamento do modelo atômico precisam ser constantemente desenvolvidos, para lidar com as novas demandas. Esse é um novo desafio a ser enfrentado nas próximas gerações de síncrotron: lidar com os dados gerados (transferir e processar) em velocidades compatíveis com a coleta dos mesmos.
Benefícios. A intensidade de uma reflexão qualquer medida no detector é diretamente proporcional à intensidade do feixe incidente (Io), e ao volume do cristal (Vcristal). De maneira contrária, quanto maior ou mais volumosa a cela unitária (Figura 1b), menor a intensidade do sinal. Ou seja:
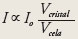
Além do mais, grandes celas unitárias e/ou cristais maiores geralmente acumulam mais desordem, o que compromete o sinal de interferência construtiva. O aumento no brilho do feixe de raios X incidentes, cada vez menores, combinado com a maior sensibilidade na detecção do sinal permitem a coleta de dados de grandes macromoléculas fracamente difratoras no estado cristalino, utilizando-se cristais menores com grandes celas unitárias (Figura 3b).
O advento de síncrotrons foi o marco histórico essencial no estudo de proteínas difíceis de cristalizar ou que produzem cristais pequenos, com grandes celas unitárias, altas quantidades de solvente e de fraco poder de difração. Dentre estas estão proteínas de membrana, capsídeos virais, complexos macromoleculares (proteína:proteína) e complexos proteína:ligante.
Nesse sentido, prêmios Nobel recentes relacionados à aplicação de cristalografia de proteínas em biologia, e cujas estruturas só puderam ser determinadas em fontes síncrotron, incluem os concedidos a Peter Agre e Roderick MacKinnon em 2003 (proteínas transmembranare transportadoras de água e íons), a Roger Kornberg em 2006 (RNA polimerase), a Ada Yonath, V. Ramakrishnan e Thomas Steitz em 2009 (ribossomo) e mais recentemente, em 2012, a Brian Kobilka e Robert Lefkowitz, pelos estudos funcionais e estruturais de receptores de membrana GPCR.
CONCLUSÕES
A ciência que se pode fazer com fontes síncrotron é única e diferente. Ela beneficia toda uma comunidade de usuários acadêmicos interessados em complexos mecanismos biológicos básicos e também usuários industriais, que priorizam a descoberta e desenvolvimento de fármacos. Nesse sentido, as perspectivas das contribuições de cristalografia de proteínas, por meio de fontes de raios X de novas gerações, são animadoras.
Atualmente, existem cerca de 130 linhas dedicadas à difração de cristais de macromoléculas biológicas, que contabilizam mais de 50% do tempo de uso de laboratórios síncroton. Essas contribuíram, e ainda contribuem, com praticamente 80% da produção anual de novas estruturas cristalográficas (Figura 4).
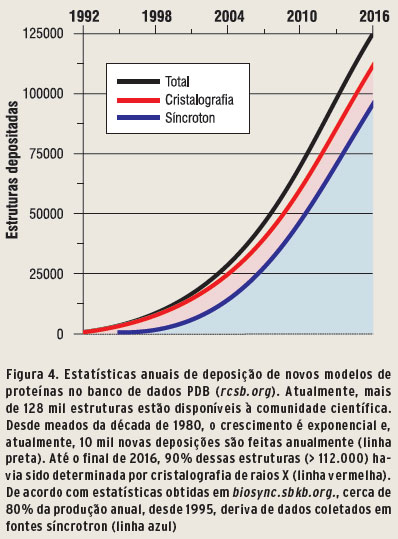
Cerca de 10.000 estruturas são depositadas anualmente, incluindo-se as redundantes. Menos de um terço das estruturas de proteínas humanas preditas (> 30.000) são conhecidas, isso sem contar eventos onde é interessante estudar a interação (complexos) entre proteínas.
Proteínas de membrana são importantes sensores do meio externo, desencadeando cascatas de respostas intracelulares vitais para a sobrevivência de uma célula e sua função organizada em tecidos e órgãos. Dessa maneira, essa classe de proteínas é de grande interesse para o desenvolvimento de fármacos. Aproximadamente 30% do genoma humano (quase 10.000 genes) codifica para essa classe de proteínas, que por sua vez são alvos conhecidos de pelo menos 50% dos fármacos que estão mercado. Todavia, proteínas de membrana são extremamente difíceis de expressar e cristalizar e seus cristais são frequentemente pequenos e frágeis, de maneira que somente uma centena de estruturas únicas são conhecidas. Assim, há ainda um longo caminho a ser percorrido na área de biologia estrutural de proteínas de membrana, e a crescente demanda por experimentos de cristalografia deve ser acompanhada pelo aumento na oferta de tempo e qualidade de feixe de raios X. Assim, a construção de novos síncrotrons de última geração (tais como o Sirius) é uma demanda a ser atendida, especialmente em regiões do planeta onde, pela escassez de ofertas, teria o potencial de contribuir fortemente com o avanço científico e econômico.
REFERÊNCIAS
1. Trinkaus, E. "Early modern humans". Annual Review of Anthropology, v. 34, n. 1, p. 207-230, 2005.
2. Sender, R.; Fuchs, S.; Milo, R. "Revised estimates for the number of human and bacteria cells in the body". PLOS Biology, v. 14, n. 8, p. e1002533, 2016.
3. Farrell, C. M. et al. "Current status and new features of the consensus coding sequence database". Nucleic Acids Research, v. 42, n. D1, p. D865-D872, 2014.
4. Mclaren, C. E.; Brittenham, G. M.; Hasselblad, V. "Statistical and graphical evaluation of erythrocyte volume distributions". The American Journal of Physiology, v. 252, n. 4, p. H857-66, 1987.
5. Blake, C. C. F. et al. "Crystallographic studies of the activity of hen egg-white lysozyme". Proceedings of the Royal Society of London B: Biological Sciences, v. 167, n. 1009, 1967.
6. Willmott, P. An introduction to synchrotron radiation: Techniques and applications. 2011.
{kind=link}
{kind=link}