
Sinalização celular em câncer
Waldemir Fernandes de Souza
Wallace Martins de Araújo
Júlio Cesar Madureira de-Freitas-Junior
José Andrés Morgado-Díaz
Organismos pluricelulares complexos (como os seres humanos) são constituídos por um conjunto de órgãos, formados por tecidos, os quais reúnem um conjunto de células especializadas para desempenharem funções específicas. O desenvolvimento desses organismos e a manutenção de suas funções vitais dependem de uma cooperação das células que os constituem, na medida em que os processos realizados por essas células têm que ser desempenhados de maneira coordenada. Para que isso ocorra, as células necessitam se comunicar umas com as outras e, a partir desta comunicação, coordenar as funções desempenhadas por cada tipo celular. Esse é o princípio da sinalização celular, onde células se comunicam através de moléculas-sinal, secretadas ou expostas em sua superfície.
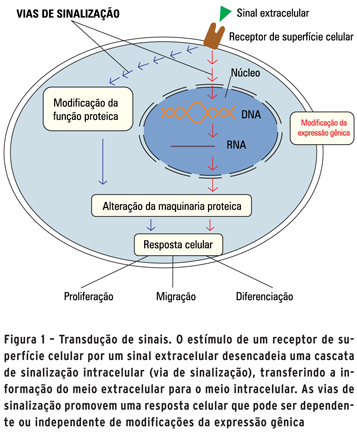
As moléculas-sinal são reconhecidas por receptores, geralmente proteínas que podem estar expostas na superfície celular ou presentes no interior das células. Esses receptores, após se ligarem às moléculas-sinal, transmitem a informação para outras proteínas presentes no interior da célula, mecanismo conhecido como transdução de sinal. Esse mecanismo pode desencadear diferentes respostas, como alteração da função de uma proteína (resposta rápida e passageira) ou modificação de expressão gênica, levando a uma alteração na quantidade de proteína dentro da célula (resposta lenta e mais prolongada). Essas modificações levam a alterações no comportamento da célula, visto que as proteínas são as responsáveis por dirigir as funções celulares (Figura 1).
A transdução de sinal ocorre através de vias de sinalização, as quais geralmente são constituídas por proteínas comprometidas com a regulação de eventos da fisiologia celular, como proliferação, migração, diferenciação celular etc. A maior parte das proteínas que participam das vias de sinalização são enzimas, biocatalizadores que atuam na aceleração das reações bioquímicas que ocorrem dentro da célula. Dentre elas encontram-se as quinases, que constituem uma das principais famílias de proteínas em mamíferos. Elas são enzimas que catalisam a transferência de grupos fosfatos de moléculas do nucleotídeo adenosina trifosfato (ATP) para outras moléculas orgânicas, como lipídeos e proteínas. Como exemplo, podemos citar as quinases reguladas por sinal extracelular 1/2 (ERK1/2, Extracellular Signal-Regulated Kinase 1/2), proteínas serina/treonina quinase que atuam na fosforilação de aminoácidos serina e treonina presentes em seus alvos proteicos; e a fosfatidilinositol 3-quinase (PI3K, PhosphatidylInositide 3-Kinase), que atua na fosforilação do fosfolipídio fosfatidilinositol presente nas membranas celulares. Outras proteínas com grande participação em vias de sinalização são as que compreendem a família das GTPases, enzimas que quando ativas atuam na hidrólise da molécula do nucleotídeo guanosina trifosfato (GTP), gerando como produto guanosina difosfato (GDP) e fosfato inorgânico. As GTPases são enzimas que ciclam entre um estado ativo ligado à GTP e um estado inativo ligado à GDP, mecanismo controlado por proteínas regulatórias. Quando ativadas, as GTPases podem se associar a proteínas efetoras regulando sua atividade. Como exemplo de GTPases podemos citar dois membros da superfamília Ras de pequenas GTPases: a Ras que atua na ativação de várias vias de sinalização, como aquelas relacionadas às proteínas ERK1/2 e PI3K; e a Rho, relacionada com a ativação de moléculas efetoras como a Rho quinase (ROCK, Rho Kinase) (1;2). A desregulação das vias de sinalização celular, causada geralmente por aumento/diminuição da atividade ou expressão de seus constituintes proteicos, pode levar ao descontrole de eventos fisiológicos e desencadear variados tipos de doenças, incluindo o câncer.
Câncer é um termo genérico usado para designar um grande grupo de doenças que pode afetar diferentes partes do organismo. Apesar da sua diversidade, o câncer apresenta características comuns: células cancerígenas (ou tumorais) apresentam desregulação nos mecanismos de controle do crescimento e proliferação, permitindo a formação de um agregado de células anormais (tumor) (3;4). Esse descontrole normalmente está associado a alterações no material genético das células, o ácido desoxirribonucleico (DNA, DeoxyriboNucleic Acid), podendo levar à desregulação da expressão de genes, os quais contêm informação para a síntese de proteínas encontradas nas células. Dessa forma, as alterações podem ser passadas para células filhas, através da divisão celular, gerando várias células anormais que formarão o tumor. As principais causas do câncer estão relacionadas à exposição a agentes externos, os chamados carcinógenos. Eles podem ser de origem física (como os raios ultravioleta da radiação solar), química (como os componentes do cigarro de tabaco) ou biológica (como a infecção de certos vírus).
Durante o desenvolvimento do tumor, vários eventos celulares podem ser desregulados. Dentre eles podemos citar o aumento do crescimento e proliferação, uma maior resistência ao mecanismo de morte celular e maior suscetibilidade a instabilidade genômica e mutações, possibilitando às células tumorais acumular alterações em seu DNA (5). Essa desregulação é um reflexo principalmente de alterações na atividade ou expressão das proteínas constituintes das vias de sinalização celular. Dessa forma, um melhor entendimento da participação das vias de sinalização celular na progressão do câncer é um passo primordial para o desenvolvimento de metodologias terapêuticas e de diagnóstico para essa doença, em particular do câncer colorretal, que ocupa o terceiro lugar em índice de incidência na população brasileira (6).
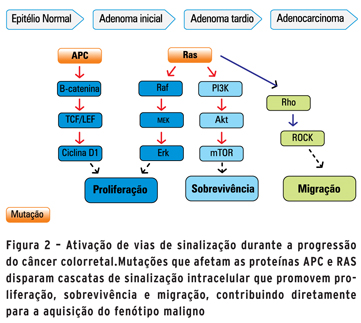
VIAS DE SINALIZAÇÃO CELULAR QUE REGULAM A PROGRESSÃO DO CÂNCER COLORRETAL O câncer colorretal (CCR) se desenvolve através de uma acumulação gradual de alterações genéticas que conduzem à transformação do epitélio normal colônico para o fenótipo maligno e invasivo. Essas alterações genéticas no epitélio intestinal podem causar mudanças histológicas e moleculares que levam a uma definição da sequência chamada de sequência adenoma-carcinoma (7). O estágio inicial da sequência adenoma-carcinoma é caracterizado por lesões aberrantes e displasias no epitélio colônico, podendo levar à formação de pólipos visíveis macroscopicamente. Esses pólipos constituem uma lesão precursora que inicia a formação do adenoma, o qual é caracterizado pela morfologia displásica e pela presença de alterações na diferenciação epitelial. A transição de um tumor benigno (adenoma) para um tumor maligno (carcinoma) deve-se ao acúmulo de mutações ao longo do estadiamento do tumor. As mutações no gene supressor tumoral APC são encontradas nos estágios iniciais da sequência adenoma-carcinoma e também em câncer hereditário, como a polipose adenomatosa familiar (FAP, Familial Adenomatous Polyposis) (8). Essas alterações no gene APC levam à perda de função da proteína APC, com uma consequente redução da degradação da proteína β-catenina, de modo a elevar seus níveis citoplasmático e nuclear nas células tumorais, conduzindo a uma desregulação da via de sinalização Wnt/β-catenina. A estabilização nuclear da proteína β-catenina é o principal mecanismo de ativação desta via, a qual é responsável pela alteração na expressão de genes alvos que conduzem a um aumento da proliferação e invasão celular (9).
A via Wnt/ β-catenina foi a primeira associada com o desenvolvimento do CCR, sendo encontrada permanentemente ativada em pacientes com FAP e em outras formas de cânceres esporádicos (10). Aproximadamente 90% do CCR esporádico mostram uma sinalização aberrante para a via Wnt, resultado de mutações frequentes em APC e também em outros genes que codificam proteínas desta via de sinalização, como β-catenina e axina. Essas mutações induzem ao acúmulo de β-catenina no citoplasma e sua translocação para o núcleo, onde interage com o fator celular-T/fator intensificador linfóide (TCF/LEF, T-Cell Factor/Lymphoid Enhancer Factor), fatores transcricionais que regulam a atividade de genes como C-MYC e MMP9, os quais estão associados a eventos da progressão tumoral, como proliferação e invasão. Essa ativação aberrante é considerada o evento inicial para a tumorigênese colorretal, levando à transformação das células do epitélio intestinal em células com capacidade proliferativa exacerbada, que culmina na formação de adenomas. Essas células podem adquirir novas mutações, as quais permitiriam a formação da metástase (11) .
Além da sinalização aberrante Wnt/β-catenina, outro evento importante no desenvolvimento do CCR é a ativação constitutiva do oncogene KRAS (Kirsten Rat Sarcoma viral oncogene homologue). Em CCR, essa forma mutante hiperativa é a segunda mais frequente em adenomas e carcinomas (12). K-RAS, um dos membros da superfamília RAS de pequenas GTPases, é responsável por mediar a transdução de sinais desencadeados pela ativação de receptores tirosina quinase presentes na superfície celular. A sinalização acontece quando moléculas-sinal extracelulares, como o fator de crescimento epidérmico (EGF, Epidermal Growth Factor), o fator de crescimento de hepatócito (HGF, Hepatocyte Growth Factor), e o fator de crescimento derivado de plaqueta (PDGF, Platelet-Derived Growth Factor), se ligam a seus receptores desencadeando sua dimerização e/ou autofosforilação em resíduos de tirosina nos seus domínios citoplasmáticos. O sinal frequentemente é repassado para a oncoproteína K-RAS, localizada na membrana celular, que em seguida pode ativar por fosforilação a via ERK1/2, causando um aumento da proliferação através da indução da expressão de ciclina D1 (13). Além disso, K-RAS também pode enviar sinais para via PI3K/Akt, que está relacionada ao aumento de sobrevivência celular.
A PI3K constitui uma família de quinase lipídica intracelular que fosforila o grupo 3'- hidroxil de fosfatidilinositol das membranas celulares. A ativação consiste em catalisar a formação de fosfatidilinositol-3,4,5-trifosfato (PIP3, PhosphatidylInositol (3,4,5)-triPhosphate) a partir de fosfatidilinositol-4,5-bifosfato (PIP2, PhosphatidylInositol (4,5)-bisPhosphate), produto que transduz um sinal por interação com proteínas de domínio homólogo a pleclestrina (PH, Pleckstrin Homology). A serina-treonina quinase Akt (também conhecida como PKB) é uma proteína efetora central do PIP3. A Akt é ativada por um duplo mecanismo regulatório que requer a translocação e ancoramento na membrana plasmática através do domínio PH sendo, logo em seguida, fosforilada em seus aminoácidos treonina 308 e serina 473 pelas quinases proteína dependente de 3-fosfoinosídeo quinase-1 (PDK-1, 3-Phosphoinositide Dependent protein Kinase-1) e proteína reguladora de mamífero para rapamicina C2 (mTORC2, mammalian Target Of RapamyCin 2) respectivamente (14). A fosfatase homóloga à tensina deletada no cromossomo 10 (PTEN, Phosphatase and TENsin homologue deleted on chromosome 10) é um fosfatase lipídica que atua na desfosforilação de PIP3, regulando negativamente a ação de PI3K, portanto assumindo um papel de supressor tumoral. A presença de PTEN mantém os níveis de PIP3 baixo, enquanto que sua ausência aumenta a concentração de PIP3, elevando a atividade de Akt. Nessa perspectiva, a ativação de PI3K por sinalização dependente de fatores de crescimento, acarreta a síntese de PIP3, que é desfosforilado por PTEN a PIP2. Na ausência de PTEN, Akt torna-se constantemente fosforilada e, portanto, mais ativa, podendo mediar a ativação de diversas proteínas que regulam a sobrevivência e o crescimento tumoral (15).
Outro grupo de proteínas que participam ativamente da progressão do CCR é o das Rho GTPases, as quais estão relacionadas com a sinalização que regula a reorganização do citoesqueleto de actina (16). As formas ativas de Rho adquirem uma mudança conformacional que permite interagir com proteínas efetoras, que na verdade desempenham a função de Rho propriamente dita. A primeira proteína efetora de Rho identificada foi a ROCK, a qual atua na indução da formação de fibras de estresse e adesão focal, através das proteínas que organizam o citoesqueleto de actina, além do controle da contratilidade celular pela miosina. Conforme já mencionado, as Rho GTPases modulam o citoesqueleto de actina, causando projeções de membrana que aumentam o potencial migratório, invasivo e metastático das células tumorais.
Nosso grupo de pesquisa no Instituto Nacional de Câncer (INCa) tem trabalhado ativamente para melhor caracterizar as vias de sinalização celular que regulam a progressão do CCR. Em trabalhos recentes, relatamos a participação das vias da PI3K/Akt, ERK1/2 e Rho GTPases regulando eventos da progressão deste tipo de câncer (17-19). Um resumo de nossos resultados está esquematizado na figura 2.
]]>
Sinalização na terapia do câncer: o tratamento racional As estratégias terapêuticas tradicionais utilizadas no tratamento do câncer empregam a cirurgia, quimioterapia e radioterapia. A escolha do tratamento ideal para um tipo de câncer geralmente se baseia em ensaios clínicos com uma grande população de pacientes, os quais apontam o melhor esquema terapêutico a ser utilizado. Contudo, durantes os ensaios clínicos podemos encontrar uma grande variação de resposta entre os pacientes, indicando que uma melhor estratificação em subgrupos poderia apontar diferentes níveis de resposta a um esquema terapêutico empregado (20). A identificação de um subgrupo de pacientes com câncer que respondem melhor a um dado esquema terapêutico pode refletir na diminuição dos custos do tratamento e aumentar os benefícios para o bem-estar dos pacientes, na medida em que se diminuem os efeitos colaterais. Nesse sentido, os estudos que visam caracterizar os biomarcadores envolvidos na progressão do câncer e os processos por eles regulados ajudam no melhor direcionamento do tratamento, na medida em que são utilizados como base para o desenvolvimento de ferramentas para o bloqueio molecular desses processos. Dentre essas ferramentas destacamos o uso dos anticorpos monoclonais e os inibidores farmacológicos, ambos atuando na inibição da atividade de proteínas específicas presentes em vias de sinalização.
Os anticorpos monoclonais são biomoléculas que se ligam a alvos específicos, geralmente receptores proteicos presentes na superfície celular, evitando a ativação ou reconhecimento desses receptores por outras moléculas. Como exemplo, podemos citar alguns dos anticorpos monoclonais aprovados pelo FDA (Federal Drug Administration), órgão de vigilância sanitária dos Estados Unidos, para tratamento do CCR metastático: o Cetuximab e o Panitumumab, ambos tendo como alvo o receptor do fator de crescimento epidérmico (EGFR, Epidermal Growth Factor Receptor). A combinação desses anticorpos anti-EGFR com esquemas terapêuticos tradicionais, que atuam no DNA das células, tem gerado bons resultados. Porém, os melhores benefícios são alcançados por uma prévia investigação acerca do status mutacional do gene KRAS, o qual codifica a proteína K-RAS, uma das efetoras ativadas pelo EGFR: caso a proteína K-RAS esteja ativada de forma constitutiva, devido à mutação em seu gene, a ativação da via de sinalização efetora continuará mesmo após a ligação do anticorpo ao EGFR. Assim, os principais órgãos de vigilância sanitária no mundo têm recomendado a investigação de mutação em KRAS antes do emprego da terapia com anticorpos anti-EGFR em pacientes com câncer colorretal metastático (21;22).
Devido à importância das proteínas intracelulares participantes das vias de sinalização, o emprego de inibidores farmacológicos destas tem sido utilizado em ensaios clínicos. Os inibidores farmacológicos são moléculas purificadas de compostos naturais ou sintetizadas em laboratórios que impedem a atividade de proteínas por alterar sua conformação ou competir por um substrato. A inibição da proteína quinase MEK (Mitogen-activated protein Kinase), pelo inibidor GSK1120212, tem contribuído para estabilização da síndrome mielodisplásica em aproximadamente 54% dos pacientes tratados, os quais apresentavam mutações nos genes KRAS ou NRAS (23). Além disso, estudos em laboratório têm mostrado recentemente que o inibidor TIC10, o qual inibe ambas as vias de sinalização Akt e ERK1/2, atua como potente agente antitumoral, sendo uma molécula promissora para análise em futuros ensaios clínicos (24). Poucos estudos têm relacionado a inibição da via de sinalização regulada pela Rho GTPase com o tratamento antitumoral. Porém, o inibidor de ROCK, Fasudil, tem sido usado no tratamento de pacientes com vasoespamos no cérebro sem causar sérios efeitos colaterais. Em estudos no laboratório, células tratadas com Fasudil apresentaram redução na sua migração e na capacidade de formar colônias não aderidas ao substrato, características indicativas de redução do potencial maligno dessas células, sugerindo que este inibidor pode ser uma ferramenta potencial para o tratamento antitumoral (2).
Esse conjunto de dados aponta a importância da caracterização das alterações moleculares no tumor para um melhor direcionamento do tratamento. Nesse sentido, o emprego de técnicas de análise em larga escala, como o microarranjo de DNA, permite avaliar alterações na expressão gênica de um grande número de amostras. Apesar de ainda apresentar um alto custo, essa ferramenta permite a identificação de possíveis biomarcadores. A partir dessa identificação, podemos direcionar melhor o tratamento do câncer, utilizando, por exemplo, anticorpos monoclonais e/ou inibidores farmacológicos para alvos proteicos específicos, os quais podem ser combinados com os esquemas terapêuticos tradicionais, objetivando uma maior eficácia do tratamento e diminuindo os efeitos colaterais. Estudos que buscam caracterizar as principais vias de sinalização que regulam a progressão de um tipo de câncer podem, no futuro, reduzir os custos para utilização das análises em larga escala, na medida em que buscam uma determinação no número de genes que precisarão ser testados. Isso pode contribuir para um tratamento mais personalizado e menos agressivo para os pacientes.
Waldemir Fernandes de Souza é biólogo pela Universidade Federal do Rio de Janeiro (UFRJ), doutor em oncologia pelo Instituto Nacional de Câncer (INCa) e atualmente é pós-doutorando do Grupo de Biologia Estrutural do Centro de Pesquisas do INCa.
Wallace Martins de Araujo é biólogo pela UFRJ, doutor em ciências morfológicas pela Universidade do Estado do Rio de Janeiro (UERJ) e atualmente é pós-doutorando do Grupo de Biologia Estrutural do Centro de Pesquisas do INCa.
Julio Cesar Madureira de-Freitas-Junior é biólogo pela UERJ, doutor em ciências pela UERJ e atualmente é pós-doutorando do Grupo de Biologia Estrutural do Centro de Pesquisas do INCa.
José Andrés Morgado-Díaz é biólogo pela Universidad Nacional de Trujillo, Peru, doutor em bioquímica pela UFRJ, com pós-doutorado pela Fundação Oswaldo Cruz (Fiocruz) e pelo National Institute of Health (NIH), EUA. Atualmente é pesquisador titular do Centro de Pesquisas do INCa, onde lidera o Grupo de Biologia Estrutural, e pesquisador nível 1D do CNPq.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Meloche, S.; Pouysségur, J. "The ERK1/2 mitogen-activated protein kinase pathway as a máster regulator of the G1- to S-phase transition". Oncogene, vol.26, p.3227-3239, 2007.
2. Leve, F.; Morgado-Díaz, J.A. "Rho GTPase signaling in the development of colorectal cancer". Journal of Cellular Biochemistry, vol.113, p.2549-2559, 2012.
3. Instituto Nacional de Câncer José Alencar Gomes da Silva. "O que é câncer?". Disponível em: <http://www1.inca.gov.br/conteudo_view.asp?id=322> (acesso em 14/10/2013).
4. World Health Organization. "Cancer". Disponível em: <http://www.who.int/mediacentre/factsheets/fs297/en/index.html> (acesso em 14/10/2013).
]]>5. Hanahan, D.; Weinberg, R.A. "Hallmarks of cancer: the next generation". Cell, vol.144, p.646-74, 2011.
6. Instituto Nacional de Câncer José Alencar Gomes da Silva. Coordenação Geral de Ações Estratégicas. Coordenação de Prevenção e Vigilância. "Estimativa 2012: incidência de câncer no Brasil". Rio de Janeiro, INCa, 2011. Disponível em: <http://www1.inca.gov.br/estimativa/2012/estimativa20122111.pdf> (acesso em: 21/10/2013).
7. Radtke, F.; Clevers, H. "Self-renewal and cancer of the gut: two sides of a coin". Science, vol.307, p.1904-1909, 2005.
8. Fodde, R.; Smits, R.; Clevers, H. "APC, signal transduction and genetic instability in colorectal cancer". Nature, vol.1, p.55-67, 2001.
9. Klaus, A.; Birchmeier, W. "Wnt signalling and its impact on development and cancer". Nat Rev Cancer, vol.8, p.378-98, 2008.
]]>10. Anastas, J.N.; Moon, R.T. "Wnt signalling pathways as therapeutic targets in cancer". Nat Rev Cancer, vol.13, p.11-26, 2013.
11. Clevers, H.; Nusse, R. "Wnt/β-catenin signaling and disease. Cell, vol.149, p.1192-205, 2012.
12. Arends, M.J. "Pathways of colorectal carcinogenesis". Appl Immunohistochem Mol Morphol, vol.21, p.97-102, 2013.
13. Wickia A.; Herrmannb, R.; Christoforic, G. "Kras in metastatic colorectal cancer". Swiss Med Wkly. vol.104, p.1-17, 2010.
14. Vivanco, I.; Sawyers, L. "The phosphtidylinositol 3-kinase-Akt pathway human cancer". Nature, vol.2, p.489-501, 2002.
]]>15. Song, M.S.; Salmena, L.; Pandolf, P.P. "The functions and regulation of the PTEN tumour suppressor". Nat Rev Mol Cell Biol, vol.13, p.283-96, 2012.
16. Vega, F.M.; Ridley, A.J. "Rho GTPases in cancer cell biology". FEBS Lett, vol.582, p.2093-101, 2008.
17. De Araújo, W.M. et al. "PI3K-Akt and GSK-3β prevents in a differential fashion the malignant phenotype of colorectal cancer cells". J. Cancer Res Clin Oncol, vol.136, p.1773-1782, 2010.
18. De Souza, W.F. et al. "Claudin-3 overexpression increases the malignant potential of colorectal cancer cells: roles of ERK1/2 and PI3K-Akt as modulators of EGFR signaling". PLoS One, vol.8, nº 9, p.e74994, 2013.
19. Leve, F. et al. "Lysophosphatidic acid induces a migratory phenotype through a crosstalk between RhoA-Rock and Src-FAK signalling in colon cancer cells". Eur J Pharmacol, vol.671, nº 1-3, p.7-17, 2011.
]]>20. Efferth, T. "Signal transduction pathways of the epidermal growth factor receptor in colorectal cancer and their inhibition by small molecules". Curr Med Chem, vol.19, nº33, p.5735-5744, 2012.
21. Schaeybroeck, S.V.; Allen, W.L.; Turkington, R.C.; Johnston, P.G. "Implementing prognostic and predictive biomarkers in CRC clinical trials". Nat Rev Clin Oncol, vol.8, p.222-232, 2011.
22. Jain, V.K.; Hawkes, E.A.; Cunningham, D. "Integration of biologic agents with cytotoxic chemotherapy in metastatic colorectal cancer". Clin Colorectal Cancer, vol.10, nº 4, p.245-257, 2011.
23. Bachegowda, L. et al. "Signal transduction inhibitors in treatment of myelodysplastic syndromes". J Hematol Oncol, vol.6, p.50, 2013.
24. Allen, J.E. et al. "Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, trail gene induction, and potent antitumor effects". Sci Transl Med, vol.5, nº171, p.1-13, 2013 ]]>